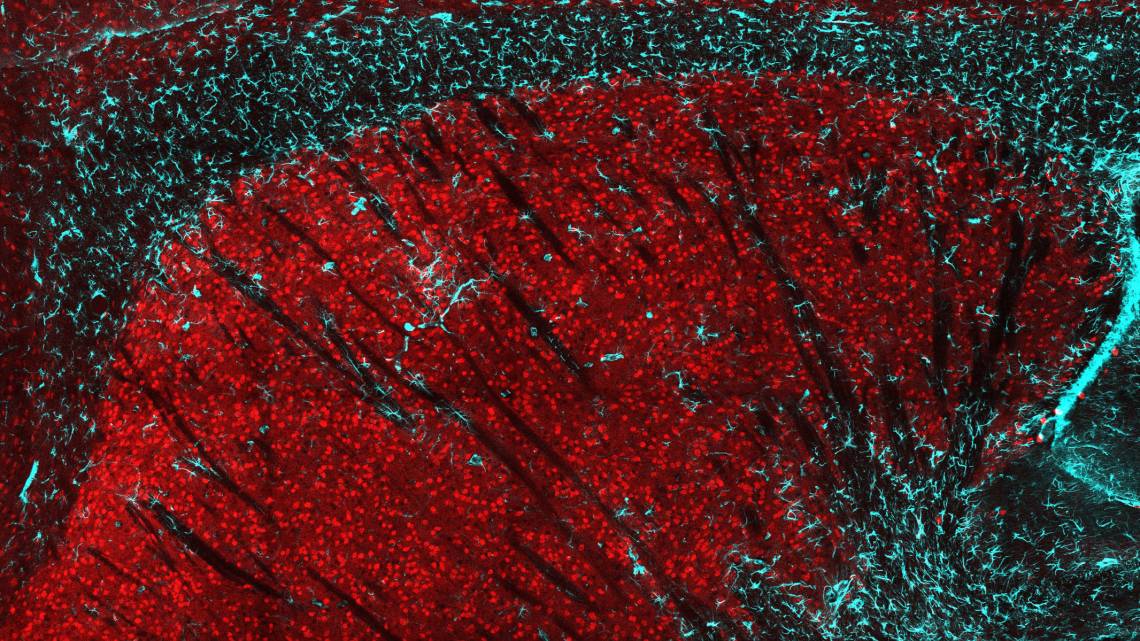
DURHAM, N.C. -- A Duke University research team has identified a new function of a gene called huntingtin, a mutation of which underlies the progressive neurodegenerative disorder known as Huntington’s Disease.
Using genetic mouse models, they have discovered that neurons in the striatum, a brain area involved in controlling movement, require the huntingtin gene for regulating the body’s movements, maintaining cell health during aging, and developing functioning connections between cells.
Addressing the gene’s role in maintaining those neural connections may provide a new avenue against Huntington’s, the researchers said.
Huntington’s Disease is an inherited neurodegenerative disorder that usually emerges in mid-life and leads to impaired motor control, dementia, and psychiatric symptoms. While the genetic basis of this lethal disease was identified more than two decades ago, there are no approved treatments yet to slow its progression or cure it.
The disease is caused by a mutation in one of a person’s two copies of the huntingtin gene. The mutation results in the production of an aberrant version of the huntingtin protein, which is toxic to neurons. Although the mutant protein is expressed throughout the body, neurons of the striatum are specifically vulnerable to its effects and degenerate as the disease progresses.
While the mutant huntingtin protein is damaging to neurons, it may also interfere with the remaining, non-mutated huntingtin’s ability to perform its normal functions.
Drugs currently being tested in clinical trials are designed to block the defective huntingtin protein, but they also end up decreasing the amount of normal huntingtin in neurons. Huntingtin is known to play several important functions in cells, but its specific role in striatal neuron health and function was not known.
“We hypothesized that the normal huntingtin gene plays a critical role in neuronal health and connectivity, and we wanted to determine what happens to striatal neurons that have had huntingtin eliminated,” said lead author Cagla Eroglu, an associate professor of cell biology and neurobiology and the co-director of Regeneration Next Initiative at Duke.
In a study appearing Tuesday in Cell Reports, the team found that deleting the huntingtin gene specifically from the striatal neurons of very young mice caused these neurons to die as the mice aged, similar to the pattern of neuron death seen in Huntington’s Disease. They also found that mice lacking huntingtin in their striatal neurons were impaired in their ability to control their movement. Importantly, this loss of movement regulation happened even before the neurons themselves started to die.
“These findings suggest that cell death itself might not be the only trigger of Huntington’s Disease symptoms,” Eroglu said.
In a healthy brain, striatal neurons control movement by communicating with other neurons through connections called synapses. The researchers found that striatal neurons lacking huntingtin formed abnormal synaptic connections, which could potentially explain the problematic motor function of the mice.
“We believe changes at the neuronal and synapse level happening before cell death are contributing to the progression of the disease,” said Caley Burrus, a PhD candidate in Eroglu’s lab and first author of the study.
It’s possible, the authors said, that therapies to address the faults at the level of the synapse may be beneficial for patients. The team is continuing the research by investigating the precise role huntingtin plays in synapse development, which might lead to specific drugs designed to target these deficits.
“Understanding the critical roles huntingtin plays in neurons is essential to designing therapies that will help individuals with Huntington’s Disease in the safest and most effective way possible,” Eroglu said.
The study was supported by grants from the National Institutes of Health (NIH RO1 NS096352 and NIH RO1 NS094754, NIH F31 NRSA 106777-01). Other authors include Spencer McKinstry, Yonca B. Karadeniz, Atesh K. Worthington, M. Ilcim Ozlu, Aditya V. Santoki, Francia Y. Fang, Annie Ma of Duke Cell Biology; Namsoo Kim and Henry Yin, of the Duke Psychology & Neuroscience; Ioannis Dragatsis at the Center for Genomics & Bioinformatics, The University of Tennessee Health Science Center, Memphis; and Scott Zeitlin of the Department of Neuroscience, University of Virginia, School of Medicine, Charlottesville, VA.
CITATION: Striatal Projection Neurons Require Huntingtin for Synaptic Connectivity and Survival, Caley J. Burrus, Spencer U. McKinstry, Namsoo Kim, …, Scott Zeitlin, Henry H. Yin, Cagla Eroglu. Cell Reports, Jan. 21, 2020. DOI: 10.1016/j.celrep.2019.12.069