Cells lining the intestinal tract form a critical barrier, protecting our bodies from the billions of bacteria living in the gut. Breaches in this barrier are driven largely by a single signaling molecule called tumor necrosis factor (TNF), elevated amounts of which are associated with inflammatory bowel diseases like Crohn’s disease and ulcerative colitis.
Drugs targeting TNF have become an effective treatment against these illnesses, but despite its clinical importance, it is still not clear what triggers an uptick in TNF levels in the gut, or how that event leads to the onset of disease.
Duke researchers now have discovered that a gene called uhrf1 acts like a kind of molecular handbrake on TNF. In the absence of uhrf1, TNF rolls out a series of pro-inflammatory and immune signals that inflame and damage the digestive tract.
“Our findings provide a new take on how inflammatory bowel diseases can emerge and develop," said Michel Bagnat, Ph.D., an assistant professor of cell biology at Duke University School of Medicine. “We already knew that genetic susceptibility could play a part, but we've found that it is not just the immune genes themselves, but also the regulation of those genes (through epigenetics), that can cause problems.”
The findings appear the week of February 16 in the Proceedings of the National Academy of Sciences.
Inflammatory bowel diseases (IBD) are a group of chronic disorders of the gastrointestinal tract that affect over 1.6 million Americans. Though the origins of these diseases are unclear, recent research has implicated a number of factors, including genetic variation, intestinal microbes, overactive immunity, and environmental exposures.
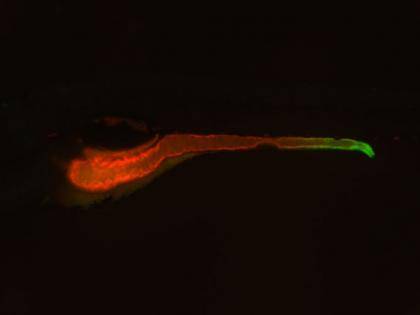
Bagnat decided to use an experimental approach called forward genetics to uncover new causes of IBD. First, his postdoctoral fellow Lindsay Marjoram used chemicals to induce mutations in their model organism of choice, the zebrafish. Because these small aquarium fish are transparent as embryos, she could easily visualize any defects in the gut as they developed.
After screening hundreds of mutants, Marjoram found several strains that displayed marked gut defects, including a thinner protective barrier and chunks of floating cellular debris.
Next, the researchers decided to narrow down their results to only the mutants involved in inflammation. Because TNF activity is a hallmark of inflammation, they created a “reporter” zebrafish that lit up green wherever the TNF gene was “turned on” in the organism. The researchers then bred the TNF reporter fish to the mutants from their initial screen to see if any of the mutated genes affected the expression of this important pro-inflammatory molecule.
The experiment yielded two big surprises. First, they found that TNF, originally thought to be produced mostly by immune cells, was also being made by the epithelial cells that line the gut. Second, they discovered that one of the mutants actually pumped up the levels of TNF being produced in the digestive tract.
After a bit more genetic investigation, the researchers found that the gene responsible was uhrf1, which is involved in an epigenetic process known as DNA methylation. Whether a particular gene is turned “off” or “on” in a given cell is determined by the presence or absence of specific chemical tags or methyl groups -- methylation -- attached to the DNA.
Uhrf1 normally acts to turn off genes that produce TNF, but when that repression is lost, those genes get turned on and TNF is manufactured and released. “You can think about it in terms of a car parked in a driveway. If you get rid of the handbrake, the car is going to start rolling,” Bagnat said.
In collaboration with Mary Goll of Memorial Sloan-Kettering Cancer Center, the researchers demonstrated that loss of uhrf1 did indeed remove methylation from the TNF gene.
Next, Bagnat turned to his Duke colleague John Rawls to explore whether the guts of the zebrafish had to be exposed to bacteria that set off TNF’s pro-inflammatory activities. When they raised the zebrafish mutants in a germ-free environment, TNF was still activated, though to a lesser extent. The results suggested that losing uhrf1’s brakes was enough to head TNF on a course for destruction, even without that extra push from intestinal microbes.
Now the researchers are trying to translate their findings to higher organisms like humans by looking for similar methylation defects in patients with IBD. Ultimately, the defects they find could provide targets for new diagnostics or therapeutics for the disease.
The research was supported by a National Institutes of Health New Innovator Award (DP2 3034656), a Bill & Melinda Gates Foundation Grand Challenges Exploration grant (OPP1108132), a Duke Multidisciplinary Fellowship in Pediatric Lung Disease (5T32HL098099-02), an F32 NRSA (DK098885-01A1), and a grant from the March of Dimes Foundation (5-FY12-93).
CITATION: "Epigenetic control of intestinal barrier function and inflammation in zebrafish," Lindsay Marjoram, Ashley Alvers, M. Elizabeth Deerhake, Jennifer Bagwell, Jamie Mankiewicz, Jordan Cocchiaro, Rebecca W. Beerman, Jason Willer, Kaelyn Sumigray, Nicholas Katsanis, David M. Tobin, John F. Rawls, Mary Goll, Michel Bagnat. PNAS, Feb. 16, 2015. DOI: 10.1073/pnas.1424089112